L’acido permanganico (HMnO4) in realtà non è isolabile come molecola singola. E’ invece ben noto il permanganato di potassio (KMnO4) che si dissolve in acqua per dare ioni permanganato MnO4– e ioni Potassio K+. L’interesse è, quindi, nella struttura geometrica ed elettronica dello ione permanganato. Tale anione ha la geometria di un tetraedro, con il manganese posto al centro e i quattro ossigeni che fungono da vertici. Nel discutere la geometria dei composti di metalli di transizione, a cui Manganese appartiene, nella letteratura chimica elementare si fa spesso uso della Teoria del Campo dei Leganti. Si veda in proposito la risposta alla domanda sugli ossidi di ferro, di cui alla pagina:
http://www.vialattea.net/esperti/php/risposta.php?num=10166
Tuttavia la teoria del campo dei leganti non funziona sempre in modo corretto e le sue risposte possono anche essere qualitativamente sbagliate. Occorre in tali casi rivolgersi a modelli più complessi e quantitativi che, attraverso opportuni calcoli quanto-meccanici, consentano interpretazioni più realistiche dei dati sperimentali a disposizione. Si ricorda, come già discusso alla pagina
http://www.vialattea.net/esperti/php/risposta.php?num=13006
che l’interpretazione delle proprietà molecolari in termini delle teorie del legame discende in modo diretto da specifiche tecniche di calcolo sviluppate per risolvere in qualche modo approssimato l’equazione di Schroedinger relativa a sistemi multielettronici, equazione che non è risolubile esattamente se non in un limitatissimo numero di casi molto semplici.
Per quanto riguarda lo ione permanganato, il modello qualitativo classico prevede una carica formale di +7|e| sullo ione manganese, che si suppone abbia perso tutti i suoi elettroni di valenza 4s23d5, ceduti interamente agli ossigeni che avrebbero carica -2|e|. In realtà, le cose vanno un po’ diversamente… In generale, la struttura elettronica di un complesso o di un ossido di un metallo di transizione è correlata alla struttura geometrica; per esempio, la geometria di un ossoanione (come il permanganato) in cui un metallo coordina 4 ossigeni potrebbe essere piano-quadrata oppure tetraedrica (con poliedri di coordinazione ideali, oppure distorti) e nei diversi casi ne discenderebbe una struttura e una distribuzione elettronica diversa. La struttura elettronica a cui compete la più bassa energia determina in ultima analisi la struttura geometrica di equilibrio (o da questa è determinata, a seconda del punto di vista). Nel caso del permanganato potremmo scegliere di descrivere la struttura elettronica come closed shell (tutti gli elettroni appaiati; sistema di singoletto, diamagnetico, con spin totale pari a zero), oppure come open shell (avente due o più elettroni spaiati; tripletto, quintetto… paramagnetico). I calcoli mostrano che lo stato energeticamente più stabile è quello di singoletto, con struttura tetraedrica (stati elettronici eccitati a più alta molteplicità di spin possono avere struttura piano-quadrata).
Uno schema delle energie degli orbitali molecolari, e della loro composizione, per lo stato di singoletto, è riportato nella figura sottostante, calcolato a livello della Density Functional Theory (DFT) usando una Hamiltoniana B3LYP, con un set base variazionale di tipo triplo Z (TZV):
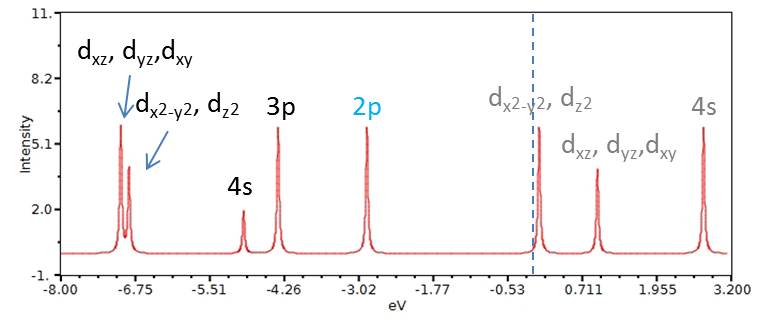
In ascissa è riportata l’energia degli orbitali (in eV, elettronvolt); in ordinata una Intensità che dipende dalla molteplicità (degenerazione) di diversi orbitali molecolari equivalenti per simmetria, che hanno la stessa energia, e dai coefficienti dei vari orbitali atomici nella specifica combinazione lineare che definisce ogni orbitale molecolare. L’area a sinistra della linea verticale tratteggiata, in azzurro, si riferisce agli orbitali occupati, mentre quella a destra della linea si riferisce agli orbitali virtuali (appunto non occupati). Gli orbitali atomici del manganese che maggiormente contribuiscono ai vari orbitali molecolari dello ione permanganato (picchi in rosso nel grafico) sono indicati in nero (in grigio se virtuali); in azzurro sono indicati gli orbitali 2p dell’ossigeno, gli unici che contribuiscono all’orbitale molecolare a circa -3 eV. Gli orbitali s e p dell’ossigeno contribuiscono in varia misura anche agli orbitali molecolari a cui partecipano i d del manganese.
Come si vede in figura, lo splitting tipico degli orbitali d del manganese dovuto al campo tetraedrico dei leganti, che prevede un’energia più bassa per i due dz2 e dx2-y2 rispetto a quella degli altri tre orbitali, si verifica solo nel caso dei virtuali; gli orbitali d occupati presentano invece un ordine diverso a causa del contemporaneo coinvolgimento degli orbitali atomici s e p dell’ossigeno (covalenza) .
Al di là degli aspetti legati allo splitting, emerge chiaramente dalla figura il ruolo giocato dagli orbitali 4s e 3d del manganese che non sono affatto vuoti nello ione permanganato come invece vorrebbe il modello ionico che, come detto sopra, assegna formalmente una carica +7|e| al catione metallico. Al contrario, questi orbitali atomici contribuiscono a orbitali molecolari che sono anche più in basso, come energia, agli orbitali a cui contribuiscono i soli 2p dell’ossigeno (a circa -3 eV).
Dallo stesso calcolo risulta una geometria tetraedrica con distanza Mn-O pari a 0.162 nm che è molto vicina al valore sperimentale (0.163 nm, quest’ultimo essendo misurato per via diffrattometrica su un cristallo di permanganato di potassio). La carica sui singoli atomi dello ione è stimabile attraverso un’analisi di Mülliken sugli orbitali molecolari: ne risulta una carica positiva di circa 0.7|e| per il manganese, e -0.425|e| per gli ossigeni (si ricordi che, nel complesso, lo ione permanganato porta una carica negativa). Un’analisi più complessa della densità elettronica calcolata con i metodi proposti da Richard Bader nel quadro della Quantum Theory of Atoms In Molecules (QTAIM) fornisce valori di 1.64|e| per la carica del manganese e -0.66|e| per la carica degli ossigeni. Questi valori (dei quali l’ultimo è da considerarsi più oggettivo di quello ricavato dalla Mülliken) sono molto distanti da quelli pari +7|e| e -2|e| di Mn e O rispettivamente, del modello qualitativo ionico.
Il colore violetto delle soluzioni contenenti lo ione permanganato (o il colore dei cristalli di permanganato di potassio) è dovuto all’assorbimento della luce del colore complementare, nella zona del giallo, intorno ai 525 nm. Il calcolo dell’energia della corrispondente transizione elettronica con il metodo Time Dependent DFT (TDDFT, uno dei metodi più efficaci nel fornire i valori delle energie di transizione a stati elettronici eccitati, a costi computazionali ragionevoli) fornisce un valore pari a 2.25 eV che corrisponde a una lunghezza d’onda pari a 550 nm, molto vicino al valore sperimentale. La transizione è quella tra gli orbitali molecolari HOMO (Highest Occupied Molecular Orbital, quelli in cui sono coinvolti i soli p degli ossigeni a -3 eV) e LUMO (Lowest Unoccupied Molecular Orbital, coinvolgenti i dz2 e dx2-y2 del manganese. Una rappresentazione grafica della transizione è data nella figura sottostante (a sinistra è rappresentato l’HOMO con i suoi orbitali p, bilobati, centrati sugli ossigeni; a destra è il LUMO con l’orbitale dz2 oltre ai p sugli ossigeni). Si tratta di una transizione di trasferimento di carica, perchè un elettrone viene trasferito da un orbitale molecolare a cui contribuiscono i soli orbitali atomici degli ossigeni, a un altro orbitale molecolare che coinvolge anche orbitali del manganese, ed è quindi un trasferimento netto di elettroni dall’ossigeno al manganese.
![]()
Il fatto che il calcolo quanto-meccanico eseguito imponendo una configurazione elettronica closed shell (a elettroni appaiati) fornisca risultati sulla geometria dello ione, e sull’energetica delle transizioni elettroniche, che sono quantitativamente in accordo con i dati sperimentali, è normalmente considerato un valido motivo per considerare corretto proprio il modello elettronico closed shell imposto! Nel caso specifico, altre configurazioni elettroniche open shell, su cui è possibile fare i calcoli, forniscono invece valori delle proprietà rilevanti che non vanno d’accordo con l’esperimento.