Questa domanda, apparentemente così lineare, copre in realtà gli ultimi 30 anni di sviluppo tecnico della biologia molecolare. Cercherò quindi di dare una risposta esauriente ma necessariamente schematica, lasciando ad ulteriori e più specifici quesiti il compito di fornire eventuali approfondimenti.
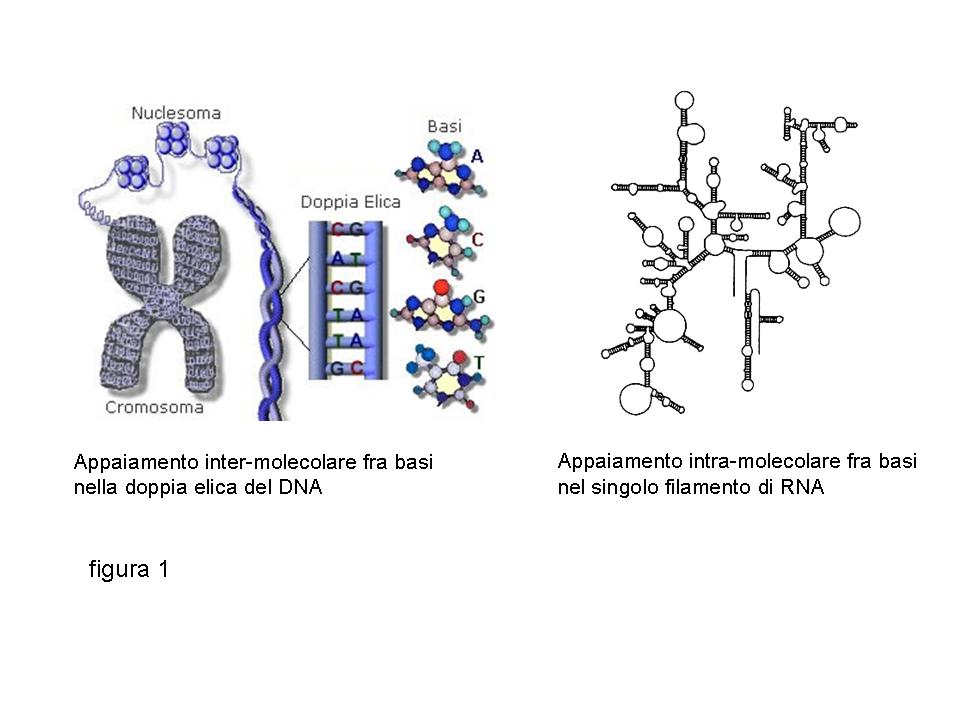
Gli acidi nucleici sono costituiti da catene di nucleotidi. Ciascun nucleotide contiene una base azotata legata chimicamente ad uno zucchero-fosfato. I due tipi di acido nucleico, DNA ed RNA, differiscono fra loro sia per la composizione in basi azotate (adenina, guanina, citosina e timina per il DNA; adenina, guanina, citosina ed uracile per l’RNA) sia per il tipo di zucchero presente (deossiribosio nel DNA, ribosio nell’RNA). Da un punto di vista strutturale, il DNA è una doppia elica contenente due catene di nucleotidi appaiate fra loro (figura 1, appaiamento inter-molecolare).
Questo appaiamento non è casuale, ma sfrutta il principio della complementarietà fra basi azotate: solo l’adenina si può trovare di fronte ad una timina e solo la citosina si può trovare di fronte ad una guanina. Questo principio di complementarietà obbligata è presente anche nell’RNA: la singola catena di nucleotidi che lo costituisce è spesso infatti resa più stabile da appaiamenti fra basi che si trovano in posizione diverse all’interno dello stesso filamento di acido nucleico (figura 1, appaiamento intra-molecolare). Questo appaiamento “forzato” è il presupposto per l’ibridazione specifica degli acidi nucleici.
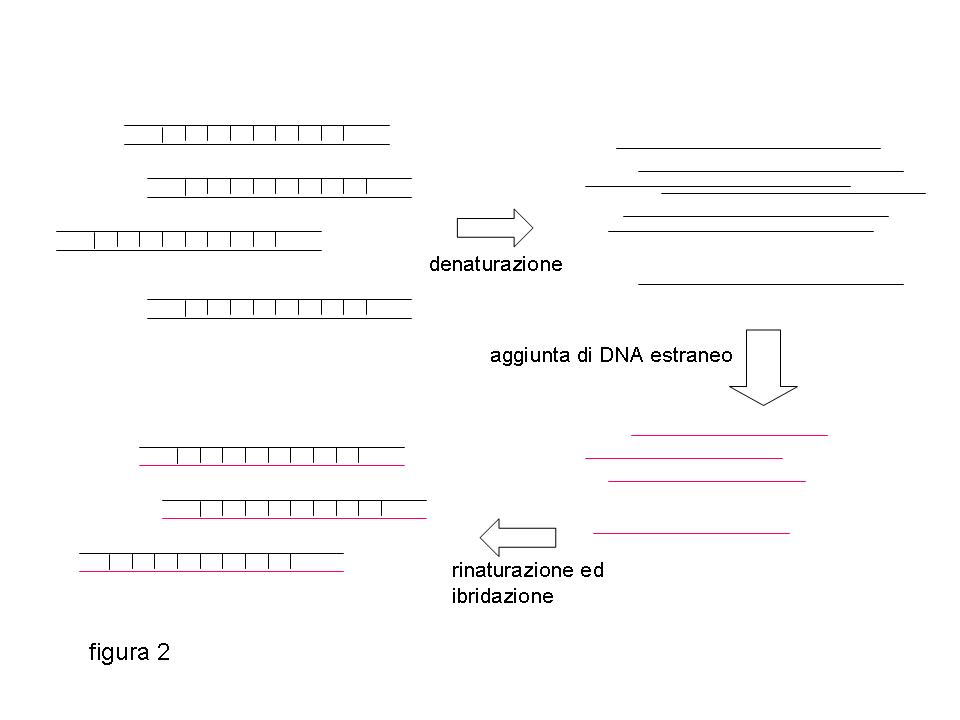
La struttura a doppia elica del DNA viene distrutta dal calore: un innalzamento della temperatura fra gli 85 ed i 95 gradi provoca la separazione dei due filamenti contenuti nella doppia elica rendendo il DNA a singola catena (denaturazione). La denaturazione è però reversibile: un successivo abbassamento della temperatura permette il riappaiamento dei due filamenti di DNA complementari in un processo noto come rinaturazione. La rinaturazione in provetta fra due molecole di acido nucleico complementari ma provenienti da fonti diverse, per esempio da organismi diversi, viene definita ibridazione (figura 2).
È importante notare come:
1. solo molecole di acido nucleico complementari possano ibridare fra loro (così come solo specifici pezzi di un puzzle possono essere incastrati fra loro)
2. i processi di denaturazione e rinaturazione descritti per il DNA sono validi anche per l’RNA
3. oltre a molecole ibride DNA:DNA, ibridi DNA:RNA sono ugualmente possibili
La tecnica nota come southern blotting prende il nome dal suo inventore, E. M. Southern, e consiste nell’isolare il DNA da un particolare organismo per poi tagliarlo con enzimi detti endonucleasi di restrizione, che lo riducono in frammenti sufficientemente piccoli da poter essere separati fra loro su di un gel d’agarosio (un pò come del terriccio passato attraverso un setaccio). Una volta separati sul gel, i frammenti di DNA vengono trasferiti su di un supporto più stabile, generalmente una membrana di nitrocellulosa o nylon, semplicemente faccendo passare grandi volumi di tampone dal gel verso la membrana (in pratica i frammenti di DNA vengono “trascinati” dal tampone). I frammenti di DNA vengono quindi “legati” alla membrana tramite calore (baking) o tramite esposizione ai raggi UV (cross-linking). La membrana, che rappresenta una copia-carbone del gel di partenza, viene quindi esposta ad una soluzione contenente un piccolo frammento di DNA radioattivo (sonda o “probe”). È importante sottolineare come il DNA venga mantenuto denaturato durante tutta la procedura. Se un DNA complementare alla sonda era presente nel campione di acido nucleico originario, la sonda vi si appaierà formando un ibrido la cui presenza può essere verificata esponendo la membrana ad una lastra autoradiografica. Questo tipo di tecnica viene solitamente usata per determinare la presenza o meno di un determinato gene nel DNA di un certo organismo.
Quando lo stesso tipo di tecnica viene applicata all’RNA, si parla di northern blotting. In questo caso, è solitamente l’RNA messaggero a dover essere isolato dal campione cellulare di partenza e corso su di un gel d’agarosio in presenza di urea, una sostanza chimica che rimuove gli appaiamenti intra-molecolari fra basi, mantenendo quindi l’RNA in forma denaturata. Nel caso del northern blotting non si effettua la digestione con endonucleasi, mentre il resto della procedura è del tutto paragonabile a quella del southern blotting. Il northern blotting viene solitamente impiegato per verificare il livello di trascrizione di un determinato gene.
Il dot blot può essere utilizzato sia su campioni di DNA sia su campioni di RNA. In entrambi i casi, gli acidi nucleici del campione di partenza vengono filtrati sottovuoto direttamente sulla membrana. In questo caso, nessuna separazione in base alle dimensioni delle molecole di acido nucleico è dunque prevista. La membrana viene quindi esposta alla sonda marcata radioattivamente. Un’eventuale ibridazione viene quindi determinata tramite autoradiografia. Poichè il dot blot richiede un numero inferiore di manipolazioni rispetto alle due tecniche precedenti, esso permette una maggiore velocità d’esecuzione e la contemporanea analisi di un maggior numero di campioni. Il dot blot è una tecnica soprattutto quantitativa che permette di determinare, per esempio, il numero di copie di un certo gene presenti in un determinato genoma o la quantità di un certo RNA cellulare.
Southern, northern e dot blotting sono tecniche classiche, sviluppate negli anni ’70 e ’80 e tuttora utilizzate ma che consentono l’analisi di un numero limitato di campioni. In anni più recenti, il completo sequenziamento del genoma umano e di quello di altre specie ha aperto la strada a quella branca della biologia molecolare nota come genomica.
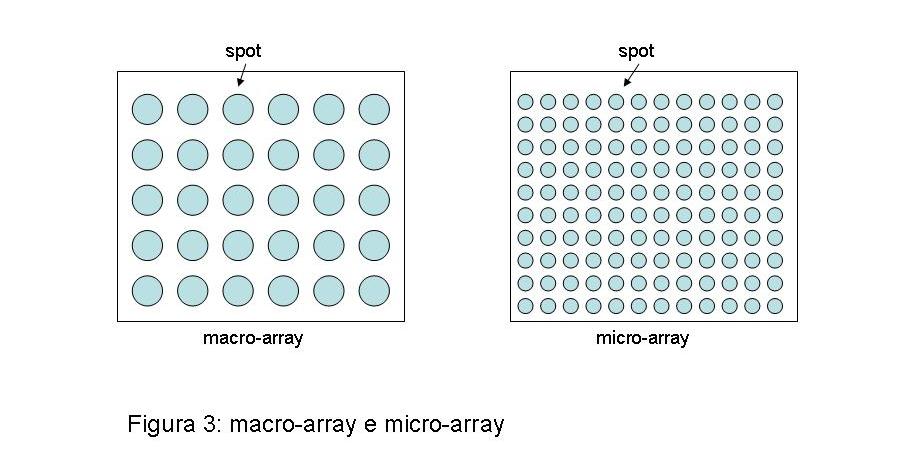
La necessità d’assegnare una funzione a ciascuno delle migliaia di geni identificati grazie alla genomica ha reso indispensabile tecniche che permettano l’analisi simultanea di moltissimi campioni. I macro- e micro-array rispondono a questa esigenza. Le due tecnologie, identiche nel principio, differiscono nel numero di geni simultaneamente analizzabili (da qualche centinaia a qualche migliaia per i macro-array; da diverse migliaia a interi genomi per i micro-array) e nel tipo di supporto utilizzato (classiche membrane di nitrocellulosa o nylon per i macro-array; supporti o “chip” in vetro o altro materiale inerte per i micro-array). Corti frammenti di acidi nucleici (“oligonucleotidi” della lunghezza compresa fra poche decine e qualche centinaia di paia di basi) corrispondenti ad un particolare tratto della sequenza dei geni presenti in un particolare tipo di cellula vengono immobilizzati in maniera ordinata e sistematica in punti precisi (o “spot”) del supporto prescelto. Il numero di geni rappresentati è quindi funzione della densità degli “spot” genici sul supporto (figura 3).
Ammettiamo si voglia ad esempio verificare l’effetto di un particolare farmaco su di un determinato tipo di cellule. Si tratteranno le cellule in questione con diversi dosaggi del farmaco, avendo cura di avere un campione con cellule non trattate (controllo positivo). Si prepareranno estratti di RNA massaggero da ciascun campione cellulare. Questi RNA messaggeri verrano poi trasformati in provetta in molecole di DNA ad essi complementari (cDNA), i quali verranno quindi marcati con isotopi radioattivi o con sostanze fluorescenti. Questi preparati saranno quindi usati come sonde ed ibridati al tipo di supporto prescelto (macro- o micro-array). L’analisi eseguita a fine ibridazione potrà rivelare che un certo numero di spot sul supporto sono ora piu’ radioattivi o piu’ fluorescenti rispetto a quanto evidenziato dal controllo positivo. Questi spot corrispondo a quei geni la cui espressione è stata in qualche modo alterata (stimolata o inibita) dal trattamento col farmaco.
Questa branca della biologia molecolare chiamata genomica funzionale è un settore in fortissima espansione, il che fa si che numerose aziende siano oggi in grado di fornire sia macro- sia micro-array già pronti per l’ibridazione, risparmiando così al ricercatore tutte le fasi di preparazione del supporto.
Venendo ora alla seconda parte della domanda, la real-time PCR (o PCR in tempo reale) è un’estensione della tecnologia di PCR classica (per una spiegazione della quale si rimanda a questa risposta già presente nell’archivio del nostro sito http://www.vialattea.net/esperti/php/risposta.php?num=7684). Mentre nella PCR classica, lo sperimentatore può verificare solo alla fine dell’esperimento il risultato dell’amplificazione, la real-time PCR consente di seguire l’evolversi della reazione in tempo reale, garantendo una maggiore flessibilità, una migliore interpretazione dei risultati ed una più semplice ed affidabile quantificazione degli stessi. Tipicamente, il parametro che viene misurato nella real-time PCR è un’aumento di fluorescenza: oltre ai due primer necessari per l’amplificazione, è presente nella reazione una sonda complementare al DNA o cDNA da amplificare. La sequenza della sonda viene scelta in modo tale che ibridizzi sul DNA/cDNA stampo all’interno delle posizioni occupate dai due primers. Questa sonda è marcata ad una estremità con un fluoroforo, una sostanza che eccitata con luce di una particolare lunghezza d’onda diviene fluorescente. All’estremità opposta a quella del fluoroforo, la sonda porta un “quencher”, una molecola che, quando è vicina al fluoroforo, è in grado d’assorbirne la fluorescenza (figura 4).
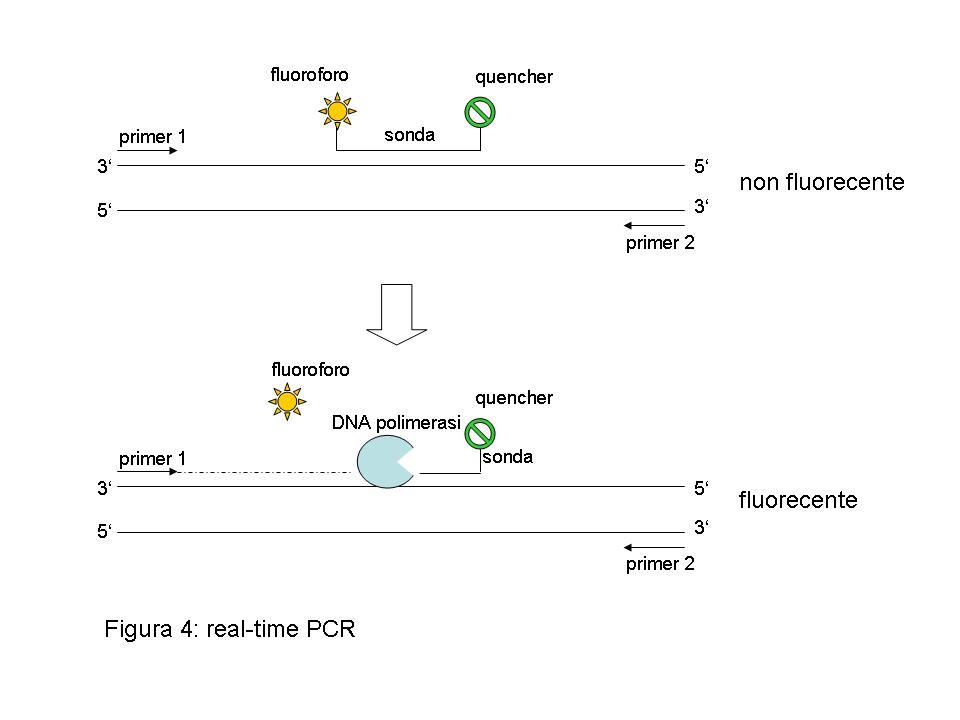
All’inizio della reazione, la sonda ibridizza al DNA/cDNA stampo ma nessuna fluorescenza può essere misurata perchè il quencher funziona da “spugna”, assorbendo l’emissione del fluoroforo. Col procedere della reazione di copiatura dello stampo a partire dal primer, però, la sonda viene gradualmente scalzata dallo stampo e degradata nei singoli nucleotidi che la costituiscono. Questo significa che ora il quencher non si trova più sufficientemente vicino al fluoroforo per assorbirne l’emissione e, di conseguenza, si assisterà ad un aumento della fluorescenza che sarà proprozionale all’efficienza dell’amplificazione dello stampo. Inoltre, se nel campione originario erano presenti solo poche molecole del DNA/cDNA stampo si assisterà ad un aumento di fluorescenza più limitato rispetto a quello ottenibile per un campione dove il DNA/cDNA stampo era presente in un maggiore numero di copie. Questo consente di trasformare le variazioni di fluorescenza misurate durante l’amplificazione in una quantificazione dell’abbondanza del DNA/cDNA stampo nel campione in analisi. Poichè diversi fluorofori sono disponibili con emissione di fluorescenza a lunghezze d’onde diverse fra loro, è inoltre possibile seguire l’amplificazione in tempo reale di diversi stampi direttamente nella stessa provetta. Sostalziamente, la real-time PCR consente una più veloce e più affidabile quantificazione dell’espressione genica, rispetto a quanto ottenibile con tecniche classiche come il northern blotting.
Per ulteriori approfondimenti (in italiano):
Benjamin Lewin “Il gene”, Zanichelli
http://www.molecularlab.it/didattica/index.asp
e in inglese:
T.A. Brown “Gene cloning, an introduction”, Chapman & Hall
E.M. Southern “Detection of specific sequences among DNA fragments separated by gel electrophoresis”, J. Mol. Biol. (1975), 98, 503-517
http://www.clinical-virology.org/anim/anim_pcr.html
http://web.ncifcrf.gov/rtp/gel/rtqpcr/default.asp
http://pathmicro.med.sc.edu/pcr/realtime-home.htm
http://opbs.okstate.edu/~melcher/MG/MGW4/MG423.html
http://www.accessexcellence.org/RC/VL/GG/southBlotg.html
http://lifesciences.asu.edu/resources/mamajis/southern/southern.html